
Monosaccharides as Useful Platform in the Synthesis of Bio-Molecules to Fight the Pathologies
Tana Lukeba Canda*, Albertina Benza Canda, Vanel Justo Bessa, Eugénio Sebastião
Departamento de Química, Universidade Agostinho Neto, Angola
*Corresponding author: Tana Lukeba Canda, Departamento de Química, Faculdade de Ciências, Universidade Agostinho Neto, Luanda, Angola
Article History
Received: November 19, 2020 Accepted: January 25, 2021 Published: January 26, 2021
Citation: Canda TL, Canda AB, Bessa VJ, et al. Monosaccharides as Useful Platform in the Synthesis of Bio-Molecules to Fight the Pathologies. Int J Pharma Sci. 2021;1(1):01‒05. DOI: 10.51626/ijps.2021.01.00001
Abstract
The development of new drugs from heterocyclic biomolecules is a challenge for scientific research. In this regard, the case of Hydantoins, tetrazoles, lactams and lactones, the most prominent of the heterocyclics offer conditions for this purpose, given their potential to confer a great diversity of bioactivities. Therefore, the development of new methods for their preparation constitutes an interesting challenge for organic chemists in general and in the area of carbohydrates in particular. In this work we report the synthesis of hydantoin, tetrazole, lactone and lactam derivatives, known as antibiotics, insecticides, analgesics, antitumor, antibacterials, antifungals and others, prepared using Schmidt, Bucherer-bergs, A.P. Rauter and Farkas reactions to monosaccharide derivatives such as keto sugars and epoxy sugars. Although they are usually reactions used for normal organic molecules, these reactions were also feasible in the monosaccharide derivatives and made it possible to obtain two tetrazole derivatives 5 and 10 with 82.9% and 48% yield, a Hydantoin derivative 3 with 62.5% yield, a lactam derivative 4 with 84.4% yield and four lactone derivatives 7a, 7b, 8a and 8b with 51%, 80%, 88.23% and 91% yield respectively. The yields were considered good in general and the methods efficient in the field of monosaccharides. The structures of these compounds were elucidated by chemical and physical tests and its IR spectra’s.
Keywords: Synthesis; Sugar derivatives; Bio-molecules
Introduction
Biomolecules are organic molecules of which are carbohydrates, vitamins, lipids, proteins and nucleic acids, 63% of which are made up of heterocyclic compounds. Among heterocyclics, hydantoins, tetrazoles, lactams and lactones stand out due to their potential as a prototype for the development of new drugs. These structural units are known to provide a wide range of naturally occurring bioactivities. Hydantoins, tetrazoles, lactams and lactones have a wide range of biological activities such as anticonvulsant, antitumor, antiviral, antibacterial, antiarrhythmic, fungicide, antibiotic, phytotoxic and antiandrogenic [1-4]. There are several methods that allow obtaining these molecules, starting from different organic compounds; however, its obtaining from carbohydrates is little reported in the literature, only a few other sugar derivatives containing this structural characteristic are described, among them is, for example, a monocyclic derivative synthesized from xylose, a bicyclic derivative obtained from D-galactose, a tricyclic derivative prepared from a hexofuranose derivative [5]; a pyrazole and tetrazole synthesized from 3-O-benzyl-1,2-O-Isopropylidene-α-D-ribo-pentodialdo-1,4-furanose [6], Synthesis of (S) -ү-hydroxymethylbutenolide from D- Mannitol for Mann [7], and others [8,9]; recently, there is also the synthesis of ү-lactones linked to sugars developed by Rauter et al. [10]. Therefore, the development of new methods for their preparation, especially in the field of carbohydrates, constitutes an interesting challenge for organic chemists. In this work we report the synthesis of hydantoin, tetrazole, lactone and lactam derivatives, using monosaccharide derivatives, such as keto sugars and sugars epoxide, as raw materials and applying the methods or reactions of Bucher-Berg [11], Schmidt [12], Farkas [13] and Rauter [10] respectively given the nature of these sugars, in order to contribute to this concern. In this context, experimental techniques adapted to classical techniques existing for these reactions were developed; in the Schmidt reaction for example, we applied a change in concentrations, which allowed us to obtain a lactam and a tetrazole, having this as the easy way to obtain lactams in keto sugars, because throughout our work we spread the developed technique by Hossein [14], recognized as an alternative to the Schmidt reaction to obtain lactams, but we did not have good results. To prepare tetrazole from epoxide, we try to obtain a nitrile derivative as an intermediate and finally we apply the Farkas [13] reaction. The heterocyclic biomolecules obtained are expected to be potential in biological activity because in their production sugars protected with isopropylidene group recognized as having biological activity were used [15-17].
Material and Methods
General procedure
The reagents and solvents used during the reactions were and when necessary, previously dried by the addition of desiccants, such as molecular sieves with a diameter of 4Å or distilled in a rotary evaporator under reduced pressure.
For the separation of organic extracts, the Büchi Rotavapor R-200 rotary evaporator was used with the V-800 vacuum controller adapted to the V-500 vacuum pump.
Column chromatography separations were performed using Fluka – 60 F254 silica gel as a stationary phase For thin layer chromatography (CCF), silica gel plates with fluorescence indicator impregnated on Merck – 60 F254 aluminium, 0.2mm thick (20×20mm) were used. After elution, the plates were observed under an ultraviolet UVGL-58 Handheld UV Lamp (λ=254nm) and developed in aqueous potassium permanganate solution. Melting points, uncorrected, were determined with the Stuart Scientific apparatus (series R000100682- 230v, ~50Hz, 75w and 1.0A [T]) and the quantities obtained in each synthesis were weighed on “Mettler Toledo” digital scales (AL 104 and PB 801). Infrared (IR) spectra were obtained using a Perkin Elmer Spectrum 1000 spectrometer. The samples were analyzed in the form of a vitreous film, deposited between cesium iodide or potassium bromide plates, using dichloromethane to dissolve the samples.
Synthesis of 1,2; 5,6-di-O-isopropylidene-α-D-ribohexofuran-3-Ulose (2) [10]
A suspension of 25mL of anhydrous dichloromethane, 5g of 4Å molecular network and 3.2g (15mmol) of PCC was prepared. Then 1.3g (5mmol) of DAG was added and the mixture was stirred for 2h at 40°C. The end of the reaction was verified by thin layer chromatography with ethyl acetate/toluene (1: 3 v/v) as an eluent. To the cooled reaction mixture was added 300ml of ethyl ether under strong stirring. After filtration of the residue, the filtrate, dark, was passed through florisil, becoming colorless and was concentrated on the rotary evaporator having obtained a white solid (2) (0.73 g, η = 56.6%). Rf = 0.84 (ethyl acetate/toluene, 1: 4 v/v). mp=78-79ºC.
Synthesis of 5- [1,2: 5,6-di-O-isopropylidene-α-D-glucofuranoside] -imidazolidine-2,4-dione (3)
Keto sugar (2) (0.5g, 1.9mmol) was added to a 100mL two-necked flask with a magnetic stirrer and reflux condenser; and 0.2g (2.9mmol) of KCN; 0.74g (7.70mmol) of (NH4) 2CO3 and 9.6ml of 50% ethanol were added. This mixture was subjected to a heating of 65oC, under stirring for 3 days (72hours). The end of the reaction was verified through the c.c.f with AcOEt/Tol 1: 4. The product was acidified with HCl (conc) to PH=2 under an ice bath. The solid was concentrated by evaporation of ethanol (aq) in the vapor route, having obtained a light yellow liquid, and it was taken to the freezer, after 2 days in the freezer, the formation of a yellow solid was verified. This yellow solid was recrystallized with a mixture of ethanol and 1: 1 hot distilled water, forming yellow crystals (3) (0.40 g; η = 62.50%); mp = 233-234oC; Rf = 0.60 AcOEt/Tol 1: 4).
Synthesis of 1,2,6,7-di-O-isopropylidene-α-D-ribo-hexopyran-3-amino-4-ulose (4)
0.18g of sodium azide were added in small portions, during 20 minutes to the solution of keto sugar 2 (0.3g) in 6mL of 36% HCl, at a temperature of 35ºC. The mixture was stirred for 3h at room temperature and the mixture was cooled to 0°C and brought to pH 10 with a 20% NaOH solution.
The NaCl precipitate was filtered and the aqueous phase was continuously extracted with chloroform for 48 h. The organic phase was dried with magnesium sulfate and a clear oily liquid was obtained. (4) (0.27g, η = 84.4%). Rf = 0.28 (ethyl acetate/toluene, 1:4v/v).
Synthesis of 1,5- (1, 2, 6, 7-di-O-isopropylidene-α-D-ribohexofurans-3, 4-yl) tetrazole (5)
0.36g of sodium azide was added in small portions during 20 minutes to the solution of keto sugar 2(0.3g) in 12mL of 36% HCl, at a temperature of 35ºC. The mixture was stirred for 3h at the end of this time, the mixture was cooled to a temperature of 0°C and brought to pH 10 with a 20% NaOH solution. The NaCl precipitate was filtered and the aqueous phase was continuously extracted with chloroform for 48h. The organic phase was dried with magnesium sulfate to obtain a yellowish oily liquid. (5) (0.29g, η=82.9%). Rf = 0.7 (ethyl acetate/toluene, 1: 4 v/v).
General procedure for the synthesis of 5, 6 –Anydro-3-O-benzyl-1,2-O-isopropilidene- α-D-glucofuranose (6) [19]
Diphenylphosphine (2, 6 equiv) was added to a solution of diol (1mmol) in benzene (17.5ml) and the mixture was stirred at rt for 15 min. After addition of activated powdered molecular sieves (3Aº. 800 g), DEAD (2,6 equiv) was added drop wise to the mixture, which was stirred at 80ºC for 48h. After filtração and solvent evaporation under reduced pressure, the residue was subjected to column chromatographic.
Starting from diol (1,33g, 4.29mmol) this procedure gave (6) (0,90g, η=72%) Rf=0.47 (Etoac/toluene 1:3 v/v); which had physical and spectroscopic data in full agreement with those given in the literature 19.
General procedure for the synthesis of the selenolactones (7a, 7b) [10]
A solution of n-butyllithium (1,6M in n-hexane 2.75mL, 4.4mmol) was added dropwise to a solution of diisopropylamine (0.62mL, 4.4mmol) in anhydrous THF (8ml) under argon at 0ºC, and the mixture was stirred at 0ºC for 25min. A solution of either phenylselenoacetic acid (2mmol) in anhydrous THF (2mL) was added dropwise at 0ºC and the reaction mixture was stirred for 1h at 0ºC. A solution of the epxide (2mmol) in abhydrous THF (3mL/g epoxide) was then added dropwise and, after stirring for 1h at 0ºC, the reaction mixture was stirred for 16h at rt. The reaction mixture was treated with 50% acetic acid (5mL) under reflux for 6h, and cooled to rt. After neutralization with a saturated NaHCO3 solution, the mixture was stracted with diethyl eter (3x10mL) and the organic phases were washed with water and dried over sodium sulfate. The lactones were isolated after solvent evaporation and purification by column chromatographic.
Synthesis of (7R) and (7S)-3-O-Benzyl-6,7-dideoxy-1,2-O-isopropylidene -7-phenylselenyl- α-D- gluco-octofuranurono-8,5-lactone (7a,7á)
Starting from (6) (171mg, 0, 585mmol), this procedure gave 7a and 7á (145mg, η=51%) ratio 7a/7a=1:1). cc eluent Etoac/toluene 1:5; Rf=0,58; which had physical and spectroscopic data in full agreement with those given in the literature10.
Synthesis of (7R) and (7S)-3-O-Benzyl-6,7-dideoxy-1,2-O-isopropylidene -7-phenylselenyl- α-D- allo-octofuranurono-8,5-lactone (7b,7b)
Starting from 6 (300mg, 1.03mmol), this procedure gave 7b and 7b´ (240mg, η=80%) ratio 7b/7b´=1:1). cc eluent Etoac/hexane 1:3; Rf=0,34; which had physical and spectroscopic data in full agreement with those given in the literature10.
Synthesis of 3-O-Benzyl-6,7-dideoxy-1,2-O-isopropylidene-α-D-gluco-octo-6-enofuranurono-8,5-lactone (8a)
Acording the general method B described by Rauter [10], starting from 7a,7a´ (50mg 0,10mmol) gave 8a (30mg, η=88,23%), CC eluent Etoac/hexane 1:4, Rf = 0,3; which had physical and spectroscopic data in full agreement with those given in the literature 10.
Synthesis of 3-O-Benzyl-6,7-dideoxy-1,2-O-isopropylidene-α-D-allo-octo-6-enofuranurono-8,5-lactone (8b)
Acording the general method B described by Rauter [10], starting from 7b,7b´ (130mg 0,3mmol) gave 8b ( 100mg, η=91%), cc eluent Etoac/hexane 1:1, Rf = 0,6; which had physical and spectroscopic data in full agreement with those given in the literature 10.
Synthesis of 3-O-benzyl-1,2-O–isopropylidene-6-deoxy -α-D-gluco-heptofuranurononitrile (9) [18]
1g(3.42mmol) of 6, 0.45g of KCN and 0.9g of MgSO4 • 6H2O dissolved in 2.27mL of distilled water were placed in a flask at room temperature for 24 hours. After that time the mixture is filtered, the filtrate is evaporated and the residue is extracted with chloroform. The organic phase is dried with MgSO4 and concentrated on the rotavapor to obtain a transparent oily liquid (9) (0.37g, η=33.9%). Rf = 0.74 (ethyl acetate/toluene, 1: 4 v/v).
Synthesis of 5- (3-O-benzyl-5-hydroxy-1,2-O-isopropylidene-α-D-gluco-furans-6-yl) tetrazole (10)
Ammonium chloride (1.47mmol, 0.08g) and sodium azide (1.27mmol, 0.11g) were added to a solution of (9) (0.37g) in 2.6ml of DMF. The mixture was heated at 100°C for 3 hours and 30 minutes. It was concentrated on the rotary evaporator and the residue was dissolved in 16ml of distilled water and extracted with 3 portions of 16ml of ethyl acetate. The organic phase was dried with MgSO4, filtered and concentrated, obtaining the final pure product, a yellowish oily liquid (10). (0.2g, η=47.6%). Rf = 0.54 (ethyl acetate/toluene, 1: 4 v/v).
Characterization
In this work, nine compounds were synthesized, they were purified and characterized, the structures of all compounds obtained was elucidated by chemicals tests, physical properties and its IR spectra’s:
Compound 2: Solid, Rf=0.84 (Etoac/toluene, 1: 4 v/v). mp = 78-79ºC. and had physical and spectroscopic data in full agreement with those given in the literature 10.
Compound 3: Solid; Rf = 0.60 AcOEt/Tol 1: 4), mp=233-234oC, IR(KBr) 1734 (C=O), 3319 (N-H),1332,8; 1317 (CO,isop) cm-1.
Compound 4: Syrup, Rf = 0.28 (Etoac/toluene, 1: 4 v/v); IR (CHCl3) 1729,60 (C=O), 3445,51 (N-H), 1377,34;1356,31 (CO, isop) cm-1.
Compound 5: Syrup, Rf = 0.7 (Etoac/toluene, 1: 4 v/v); IR (CHCl3) 1732,49;1716,23; 1683,31;1558,56;1540,33;1506,74 (C=N), 1247,49;1171,84;1035,17 (N-N=N), 1382,54 (CO, isop) cm-1.
Compound 6: Syrup, Rf = 0.47 (Etoac/toluene 1:3 v/v); which had physical and spectroscopic data in full agreement with those given in the literature 19.
Compound 7a: Syrup, Rf = 0,58; which had physical and spectroscopic data in full agreement with those given in the literature 10.
Compound 7b: Syrup, Rf = 0,34 Etoac/hexane 1:3; which had physical and spectroscopic data in full agreement with those given in the literature 10.
Compound 8a(syrup) and 8b(solid): which had physical and spectroscopic data in full agreement with those given in the literature 10.
Compound 9: Rf = 0.74 (Etoac/toluene, 1: 4 v/v), IR (CHCl3) 2257cm-1 (CN), 3446,56 (OH), 1506,93;1497,19 (C=C), 1373,99 (CO,isop) cm-1.
Compound 10: Rf = 0.54 (Etoac/toluene, 1:4v/v); IR (CHCl3) 1733,43;1683,80;1558,59; 1540,38 (C=N), 3566,25 (N-H), 3446,56 (OH), 1506,93;1497,19 (C=C), 1251,64;1215,90;1163,98 (N-N=N), 1373,99 (CO, isop) cm-1.
Results
Physical and spectroscopic data for the compounds are given in Table 1.
Table 1: Yield, physical properties and IR spectra for Compounds 3, 4, 5, 7a, 7b, 8a, 8b, 9 and 10.
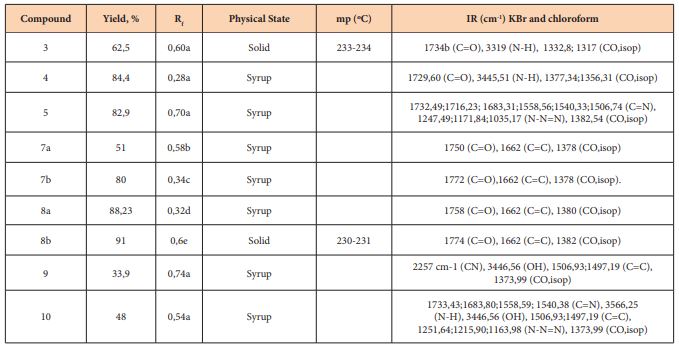
a. 1:4 Ethyl acetate/toluene; b. 1:5 ethyl acetate /toluene, c.1:3 ethyl acetate/hexane, d. 1:4 ethyl acetate/hexane; e. 1:1 ethyl acetate/hexane.
Discussion
Synthesis of Hydantoin, lactam and tetrazole from keto sugar derivative.
Keto sugar 24 was prepared by the oxidation of commercial diacetone glucose (DAG) 1 with pyridinium chlorochromate in 64% yield. Applying the Butcherer-Bergs procedure11 to keto sugar 2 using KCN and (NH4)2CO3 in Ethanol, we obtained the Hydantoin derivative 3 in 62,5% yield. On the other hand, reaction of Keto sugar 2 with the system NaN3/HCL aq, according to Schmidt´s procedure 12, afforded the lactam derivative 4 in 84,4 % yield and using the same system in excess to the keto sugar 2 it was possible to obtain the expected tetrazole 5 in 82,9 % yield. In general, this reaction has been a way to obtain amides from ketones, obtaining tetrazoles has been possible by playing with the amounts of hydrazoic acid, regarding the mechanism, the formation of tetrazoles occurs as a result of two competitive reactions: reaction of hydrazoic acid and water with aminocarbonium ion12 So it was a good strategy for us obtaining lactams and tetrazole derivatives easily, from keto sugars (Figure 1).
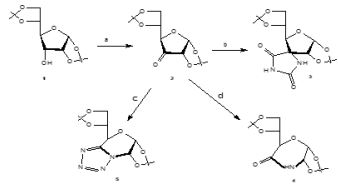
Scheme 1: Synthesis of Hidantoin, lactam and tetrazole deivatives from keto sugar 2.
a) PCC, CH2Cl2, mol. Sieve 4Aº, 40ºC, 1h; b) KCN, (NH4)2CO3, EtOH 50%,65ºC c) NaN3/HCl aq excess, 3h ; d) NaN3/HCl aq 3h.
The IR spectrum of 3 contained a carbonyl band at 1734cm-1 and the amid N-H band at 3319cm-1 and the compound 4 was confirmed for its IR spectrum with carbonyl band at 1729,60cm-1 and N-H band at 3445,51cm-1. Tetrazole 5 was confirmed by its IR spectrum which contained bands at 1732,49cm-1; 1716,23cm-1;1683,31cm-1; 1558,56cm-1; 1540,33cm-1 and 1506,74cm-1 associated at axial deformation of C=N bond of the Tetrazole ring. Chemically, the carbonyl group in these compounds was confirmed with the test of 2,4-dinitrophenyl hydrazine solution (Brady´s reagent) and the nitrogen atom by qualitative elementary analysis 20. In this reaction we used a keto sugar protected with isopropylidene and benzyl groups, we observed that these groups did not influence, sterically, the formation of the desired products.
Synthesis of lactone and tetrazole derivatives from sugar epoxide
According the procedure reported by Rauter [10], we prepared unsaturated lactone derivatives 8a, 8b in 88,23%, 91% yield respectively, by selective oxidation of the epimeric mixture of 7a,7b with hydrogen peroxide in the presence of catalytic amounts of acatic acid at 0ºC. Transformation of sugar epoxides 6a, 6b10 with the dianion of phenylselenoacetic acid afforded the epimeric α- phenylseleno lactones 7a,7b in 51%, 80% yield, respectively. The compounds 8a, 8b and 7a, 7b was confirmed by chemicals tests and its IR spectrum that show bands at 1750cm-1 (C=O) and 1772cm-1 (C=O) respectively and comparing with the literature 10. Opening the sugar epoxide 6 with KCN/MgSO4.6H2O in water 18 gave the nitrile derivative 9 and treatment of 9 with NaN3/NH4Cl in DMF, applying the Farkas´s method 13. Allowed to obtain the tetrazole derivative 10 in 48% yield (Figure 2). This method offers an important strategy in obtaining tetrazoles from epoxides since there are few examples of this case in the literature.
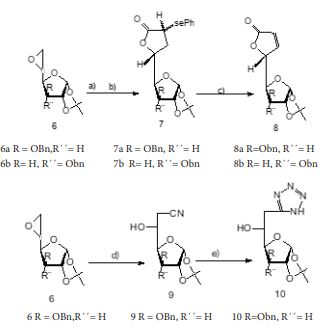
Scheme 2: Synthesis of tetrazole and lactone derivatives from sugar epoxide 6.
a) PhseCHR´´CO2H,LDA,THF,0ºC; b) CH3COOH 50%, reflux. c) H2O2 30%,CH3COOH(cat.), 0ºC ; d) KCN /MgSO4.6H2O, rt; e) NaN3/NH4Cl, DMF, 100ºC, 3h 30.
The structure of the compound 9 and 10 was also confirmed via chemicals tests and it IR spectrum which contained characteristics bands at 2257cm-1 (CN), 3446,56 (OH), 1506,93;1497,19 (C=C), 1373,99 (CO,isop) and 1733,43cm-1:1683,80cm-1; 1558,59cm-1 and 1540,38cm-1 associated at axial deformation of C=N bond of the Tetrazol ring and also bands at 3446,56cm-1 (OH), 3566,25cm-1 (N-H) respectively. Chemical tests were also made to identify the CN group and the presence of nitrogen according to the literature 20.
Conclusion
In this work, the procedures used to obtain heterocyclic biomolecules were efficient and excellent in monosaccharides because they were provided with good performance without difficulties, although they are usually applied in normal organic molecules. However, in the Schmidt reaction, we applied a change in the concentrations of the NaN3/HCL aq system, which allowed us to obtain a lactam and a tetrazole derivatives, having considered this as the easy way to obtain this compounds from keto sugars, because throughout we tested the technique developed by Hossein 14, recognized as an alternative to the Schmidt reaction to obtain amides, but we did not have good results. Based on the results obtained, we believe that the use of keto sugars and sugars epoxide in these reactions or procedures is a safe way to obtain sugar derivatives from these important heterocyclic compounds, serving their purpose in medicine. Considering that the monosaccharides with isopropylenic groups confer important biological activities [15-17], we believe that linked to the tetrazole, hydantoin, lactam and lactone units, of recognized bioactivity, we expect that the derivatives synthesized by us in this work will exhibit notorious biological activities; hence the interest in determining its bioactivity in future works.
Acknowledgment
The authors wish to thank the Angolan Ministry of Science and Technology for the financial support.
Conflict of interest
The author has no conflict if interest.
References
- Katrizky AR (2004) Introduction: Heterocycles. Chem Rev 104(5):2125-2126.
- Biljama S, Radoslav ZP (2012) Synthesis of novel 5-(Alk-3-Enyl)-Hydantoins. Sciforum.
- Batool A, Rezazadeh S (2012) A novel approach for the synthesis of 5-substituted-1H-tetrazoles. J Braz Chem Soc 23(12):2197-2203.
- Amelia P Rauter, José A Figueiredo, Isabel Ismael, Maria S Pais, António G Gonzalez, et al. (1987) Synthesis of α-Metoylene-γ-Lactones in Furanosidic Systems. J Carbohydr Chem 6(2):259-272.
- Amelia P Rauter, Hans Weidmann (1982) Reaktionen der D‐Glucuronsäure, XVI Versuche zur Verzweigung von Hexofuranurono‐6,3‐lactonen; Synthese von 5‐Desoxy‐1,2‐O‐isopropyliden‐5‐C‐methylen‐α‐D‐xylo‐ hexofuranurono‐6,3‐lacton. Liebigs Annalen der Chemie 12:2231-2237.
- Gamboa CSD (2013) Obtenção de aneis heterociclicos em derivados glucidicos, tese de Mestrado. Univ Beira Interior.
- Portero JR (1998) Synthesis de bis-α,β-butenolides Enantiopuras com simetria C2, Memória, UAB, Bellaterra, Barcelona;Spain.
- Nogueira CM, Bárbara R Parmanhan, Patrícia P Farias, Arlene G (2009) A importancia crescente dos carbohidratos em quimica medicinal. Revista Virtual de Quimica 1(2):149-159.
- Pinheiro JMA (2006) Preparação de novos pseudo C-nucleosídos e sua actividade biológica. Universidade Beira interior, Portugal.
- Rauter AP, Figueiredo J, Ismael M, Canda T, J Font, et al. (2001) Efficient synthesis of –α,β-unsaturated-ү-lactones linked to sugars. Tetrahedron Assymmetry 12(8):1131-1146.
- B Smit, Pavlovic RZ (2012) Synthesis of Novel 5-(Alk-Enyl)- Hydantoins. University of Kragujevac pp. 1-5.
- Koldobskii GI, Ostrovskii VA, Gidaspov BZ (1975) Solomon Aronovich Giller. Chemistry of heterocyclic compounds 11(6):626-663.
- Szabó IF, Farkas I, Somsák L, Bognár R (198) Acta chimica Academiae Scientiarum Hungaricae 106: 61.
- Eshghi H (2006) Ferric hydrogensulfate catalyzed Schmidt reaction of ketones to amides under solvent-free conditions, Journal of Chinese chemical Society 53:987-990.
- Fiezer LF, Fiezer A (1965) Química Orgânica. 3rd Ed, Tradução Espanhola. Mexico; 1965: 452.
- Goi A, Bruzzese T, Notarianni AF, Riva M, Ronchini A (1979) Arzneim-Forch 29: 986
- Postel DG, G Ronco I, Van Lenmene PPM, Villa PZ (1994) Patente Francesa 2: 694.
- Cyorgydeak Z, Pelyvás IF (1997) Monosaccharide Sugars. Academic Press pp. 33.
- Rauter AP, MJ Ferreira, J Font, A Virgili, M Figueredo, et al. (1995) Synthetic Fungicidal, Unsaturated-ү-lactones attached to furanosidic system. Configurational determination by Nuclear overhauser effect. J Carbohydr Chem 14: 929-948.
- Shriner RL, Hermann CKF, Morrill TC, Curtin DY, Fuson RC (1997) The Systematic Identification of Organic Compounds. 7th Ed. John willey & son; USA.
