
Jaw Keratocysts and Sotos Syndrome
Jin Fei Yeo and Philip McLoughlin*
Faculty of Dentistry, National University Centre for Oral Health Singapore, Singapore
*Corresponding author: Philip McLoughlin, Discipline of Oral & Maxillofacial Surgery, Faculty of Dentistry, National University Centre for Oral Health Singapore, 9 Lower Kent Ridge Road, Singapore
Article History
Received: June 05, 2021 Accepted: June 14, 2021 Published: June 15, 2021
Citation: Yeo JF, McLoughlin P. Jaw Keratocysts and Sotos Syndrome. Int. J. Orl. Health. 2021;1(2):31‒32. DOI: 10.51626/ijoh.2021.01.00007
Abstract
Sotos syndrome, described by Sotos et al. [1], is characterized by excessive growth during childhood, macrocephaly, distinctive facial appearance and learning disability. The disorder is largely caused by mutations or deletions in the NSD1 gene. The typical facial gestalt includes macrodolichocephaly with frontal bossing, front-parietal sparseness of hair, apparent hypertelorism, down slanting palpebral fissures, and facial flushing. This paper discusses a case of Sotos syndrome in an adolescent male with multiple odontogenic keratocysts in his jaw bones, a previously unreported oral manifestation, out with a syndromic context.
Introduction
Sotos syndrome (Cerebral gigantism) is a rare genetic childhood overgrowth condition described in 1964 by Sotos et al. [1]. This syndrome is characterized by excessive growth during childhood, macrocephaly, distinctive facial appearance and learning disability. The disorder is caused by mutations or 5q35 microdeletions in the NSD1 gene. Typical facial features include macrodolichocephaly with frontal bossing, fronto-parietal sparseness of hair, apparent hypertelorism, down slanting palpebral fissures, and facial flushing2. While oral manifestations that have been reported in Sotos syndrome include highly arched palate (>90% patients), precocious dentition (>50% patients), and mandibular prognathism (>80% patients) [2]. A single previous case with multiple odontogenic keratocysts (OKCs) has been reported, this being in association with Gorlin-Goltz syndrome [3]. We believe, however, that here is the first case of Sotos syndrome co-existing with multiple jaw keratocysts in non-syndromic isolation.
Case Report
A 17-year-old Indonesian male was referred for consultation, in June 2018, due to multiple swellings in both maxilla and mandible, which had been slowly enlarging for over a year. More recently he felt a subjective decrease in sensation of his lower lip. He was a known case of Sotos Syndrome, diagnosed clinically during childhood, and confirmed locally by genetic testing. He was allergic to Septrin, but had no other significant medical or family history.
On examination, he was a generally healthy and rather active patient. He was very tall with macrocephaly, large hands and feet, and hypertelorism with slightly down-slanted eyes. He also suffered ASD/autism and mild mental hypo development with noticeable speech impairment. On clinical assessment he was found to have a number of unerupted teeth, with fluctuant expansion of the maxilla and mandible at those sites.
Plain radiographs revealed multiples cystic lesions associated with the impacted maxillary wisdom teeth, lower left third molar, and the impacted right mandibular canine. These cysts were variously loculated, with well-defined scalloped radiologic margins, and were causing displacement of the associated teeth. (Figure 1). The maxillary cysts were occupying almost the whole of the maxillary sinuses.
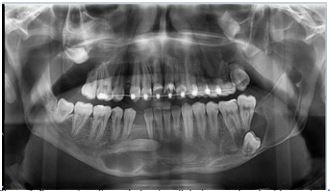
Figure 1: Panoramic radiograph showing all the impacted teeth with associated cystic lesions.
Biopsy of the anterior mandibular lesion with local anesthetic gave a histological diagnosis of odontogenic keratocyst (OKC) (Figure 2). The body of evidence favoured multiple keratocystic lesions, so under general anesthetic, all cystic masses and associated teeth were removed with the careful application of Carnoy’s solution to the bone cavities. Both inferior dental nerve bundles were preserved and the patient retained sensation to his lower lip postoperatively. Histopathology confirmed a diagnosis of OKC for all the enucleated cysts. Bone infill was reported complete at 9 months, and no cyst recurrence has occurred within the current limited observation period. The patient remains under local six monthly review, dictated by current Covid travel restriction.
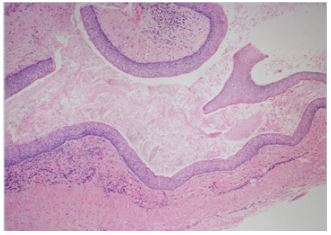
Figure 2: Photomicrograph showing part of the anterior mandibular cyst, having a cyst wall lined by uniformly parakeratinized stratified squamous epithelium, with prominent palisaded basal columnar or cuboidal cells, and a corrugated surface with abundant keratin squames within the lumen of the cyst. H & E x 100.
Discussion
Sotos syndrome, also known as “cerebral gigantism” was first described by Sotos et al. [1], with hundreds of cases, and several clinical studies, subsequently reported, although few within the Oral and Maxillofacial literature [1,4,5].
The cardinal features are pre- and postnatal accelerated somatic growth, characteristic facial appearance (i.e., macrocephaly, prominent jaw, and a high hairline with sparse hair growth), advanced bone age, and cognitive developmental delay [2,4,5]. Additional features that may be present include neonatal hypotonia, seizures, scoliosis, strabismus, congenital heart defects, and malignancies [4]. Prior to genotype identification, the diagnosis of Sotos syndrome had been based primarily on the presence of the characteristic facial appearance in a large child. However, although the combination of craniofacial features is distinctive, individual components are nonspecific, and diagnosis was occasionally a challenge [4].
Sotos syndrome is caused by mutations or 5q35 microdeletions in the NSD1 gene; a 2.2-Mb chromosome 5q35 microdeletion, encompassing NSD1, is the major cause of Sotos syndrome, accounting for 76% of cases, with intragenic NSD1 mutations identified in a minority of cases [5,6]. The original recognised oral nd craniofacial manifestations in Sotos syndrome are well described 2,4,7, so far there are no reports in the literature of multiple jaw cysts having been directly associated with the syndrome in isolation.
It is well known that multiple odontogenic keratocysts are a feature of Gorlin-Goltz syndrome, latterly re-titled Nevoid Basal Cell Carcinoma Syndrome (NBCCS), which account for about 5% of all OKCs [8,9]. However, in the case here reported, there were no other diagnostic criteria of Gorlin-Goltz syndrome unlike the recent case of Lata et al. [3]. There is a known association of OKCs with mutation or inactivation of the PTCH1 gene (mapped to chromosome 9q22.3 region) which activates the SHH signaling pathway resulting in aberrant cell proliferation of the OKC epithelium [10]. This may represent a common genetic characteristic of Sotos and Gorlin-Goltz syndromes, or incomplete genetic penetrance of the latter.
While multiple odontogenic keratocysts have been reported in Noonan syndrome [11], and in Ehlers-Danlos syndrome [12], this case report is the first occurrence of multiple OKCs in a patient with Sotos syndrome. Genetic profiling methods have potential to reveal further genotype-phenotype associations, and interactions of the NSD1 and PTCH1 genes, determining the relationship between growth modifications in Sotos syndrome and multi-cyst development.
Conflict of Interest
We have no conflicts of interest.
Ethics Statement/Confirmation of Patient’s
Permission
Ethics Statement/Confirmation of Patient’s Permission
References
- Sotos JF, Dodge PR, Muirhead D, JD Crawford, NB Talbot (1964) Cerebral Gigantism in Childhood. A syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder. N Engl J Med 271: 109-116.
- Gorlin RJ, Pindborg JJ, Cohen MM. Syndromes of the Head and Neck. 2nd Ed. Oxford; 1976: 64-66.
- Lata J, Kaur J (2020) A Unique Case of Gorlin-Goltz Syndrome with Associated Sotos Syndrome. Ann Maxillofac Surg 10(1): 232-237.
- Cole TRP, Hughes HE (1994) Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 31(1): 20-32.
- Lane C, Milne E, Freeth M (2017) Characteristics of Autism Spectrum Disorder in Sotos Syndrome. J Autism Dev Disord 47(1): 135-143.
- Douglas J, Hanks S, Temple K, Davies S, Murray A, et al. (2003) NSD1 Mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 72(1): 132-143.
- Kotilainen J, Pohjola P, Pirinen S, Arte S, Nieminen P (2009) Premolar hypodontia is a common feature in Sotos syndrome with a mutation in the NSD1 gene. Am J Med Genet A 149A (11): 2409-2414.
- Howell B, Caro M (1959) The basal-cell nevus: Its relationship to multiple cutaneous cancers and associated anomalies of development. Arch Dermatol 79(1): 67-80.
- Gorlin R, Goltz R (1960) Multiple nevoid basal cell epithelioma, jaw cysts and bifid rib. A syndrome. N Eng J Med 5(262): 908-912.
- Ohki K, Kumamoto R, Ichinohasama R, Sato T, et al. (2004) PTC gene mutations and expression of SHH, PTC, SMO, and GLI-1 in odontogenic keratocysts. Int J Oral Maxillofac Surg 33(6): 584-592.
- Connor JM, Evans DA, Goose DH (1982) Multiple odontogenic keratocysts in a case of the Noonan syndrome. Br J Oral Surg 20(3): 213-216.
- Starzynska A, Adamska P, Adamski L, Sejda A, Wychowański P, et al. (2021) Multiple odontogenic keratocysts in Ehlers-Danlos syndrome: a rare case report. BMC Oral Health 21: 107-113.
