
A Case of Alcohol Use Disorder, Osmotic Demyelination Syndrome after Recovery from Wernicke Encephalopathy Induced SIADH
Richmond Ronald Gomes*, Sayeda Noureen
Ad-din Women’s Medical College Hospital, Bangladesh
*Corresponding author: Richmond Ronald Gomes, Associate Professor of Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh
Article History
Received: June 30, 2021 Accepted: July 09, 2021 Published: July 12, 2021
Citation: Gomes RR, Noureen S. A Case of Alcohol Use Disorder, Osmotic Demyelination Syndrome after Recovery from Wernicke Encephalopathy Induced SIADH. Int J Neuropsy Beh Sci. 2021;2(2):48‒52. DOI: 10.51626/ijnbs.2020.02.00012
Abstract
It has been suggested that the syndrome of inappropriate antidiuretic hormone secretion might occur in Wernicke’s encephalopathy. We present a case report of a man with a long history of alcohol use disorder who was found to have both Wernicke-Korsakoff Syndrome (WKS) and Osmotic Demyelination Syndrome (ODS) on MRI due to attempted rapid correction of hyponatremia secondary to syndrome of inappropriate ADH secretion (SIADH). After a discussion of the case, we review the literature of the clinical manifestations, diagnostic criteria, and prognostic indicators of functional outcomes for both WKS and ODS. We compare and contrast the two diseases individually and simultaneously, as occurred in our patient.
Keywords: Alcohol use disorder; Wernicke-korsakoff syndrome; Osmotic demyelination syndrome; hyponatremia; SIADH
Introduction
Wernicke-Korsakoff syndrome (WKS) is a spectrum of serious neurologic disorders caused by to vitamin B1(thiamine) deficiency, usually in the context of chronic alcohol use disorder and poor nutrition [1]. Wernicke’s encephalopathy (WE), the acute form the disease, is characterized by the classic triad of nystagmus, encephalopathy, and ataxia [2-5]. and Osmotic Demyelination Syndrome (ODS) is a demyelinating disorder that typically occurs with the rapid correction of serum sodium in hyponatremic patients occurs in patients with chronic alcoholism, liver disease and malnutrition [6]. Previous postmortem studies suggested that pathological lesions of WE were found at 30-40% of ODS patients [7,8]. These two conditions have been reported rarely in patients without alcoholism [9-12]. However, little is known about the radiological changes combined with CPM and WE. The interval time from WE to CPM development also remains unclear. Diagnosis of both WKS and ODS requires a high index of suspicion. Though overlapping in some aspects of clinical manifestations, the prognosis and functional outcomes can differ between the two diseases so it is important to distinguish between the two processes. It has been suggested that the syndrome of inappropriate antidiuretic hormone secretion might occur in Wernicke’s encephalopathy [13,14]. However, the authors are unaware of published evidence for this although Ebels [14] mentions he has such evidence in a patient. A case is reported in which reversal of the syndrome of inappropriate antidiuretic hormone secretion occurred with intravenous vitamin B complex.
Case Report
A 65-year-old businessman, ex-smoker, not known to have diabetes, hypertension, obstructive airway disease or coronary artery disease, presented to our emergency room with 1 week history of worsening confusion, difficulty walking, and short-term memory loss. He was admitted for 22 days in a local hospital prior presenting to us with the diagnosis of vomiting induced severe symptomatic hyponatremia (serum sodium 106mmol/L). There he was treated with 3% sodium chloride without any significant clinical improvement. Sodium was only partially corrected on discharge (112mmol/L). Initial CT scan of brain at local hospital was non conclusive. He was alcoholic for last 20 years. Alcohol intake was estimated to be approximately 3-5 glasses per day; last drink was reportedly 5 days prior to presentation to local hospital.
On neurological examination, there was severe truncal ataxia such that the patient could not sit up in bed.
He was drowsy with poor concentration and recall, tangential conversation with confabulation. Pupil size and light reflexes were normal. Muscle stretch reflexes were normal in four extremities. There were no Babinski’s signs. Examination of ocular movement and cerebellar function was impossible. There were no stigmata of chronic liver disease or evidence of acute liver failure. He was apyrexial without meningism. There was no evidence of fluid depletion or cardiac failure but there was slight pitting oedema at the ankles. His pulse was 95/min, regular of good volume and his blood pressure 160/85mmHg. Mild pallor was present, jaundice was absent.
Initial work-up showed no abnormalities in the complete blood count, random blood sugar, TSH, Vitamin B12, HbA1c, urinalysis, chest x ray, USG of whole abdomen and EEG. She tested negative for HIV, and Syphilis. Lumbar puncture revealed normal protein, glucose, ADA (adenosine deaminase) and negative gram stain and culture. Serum electrolyte revealed sodium 115mmol/L (normal 135-145mmol/L), potassium 4.5mmol/L (normal 3.5-5.5 mmol/L). There was low blood urea (11.5mg/dl, normal 16-48mg/dl), low normal serum creatinine 0.66mg/dl (normal 0.6-1.2 mg/dl), a low serum osmolality (246mmosm/kg, normal-275-295mosm/kg) and a high urinary osmolality (507mosm/kg, normal-60-1400mosm/kg). Spot urinary sodium was 116 mmol/day. A short tetracosactrin (Synacthen) test was normal. So diagnosis of SIADH as a cause for hyponatremia was established.
Liver function tests revealed alanine aminotransferase (ALT) level of 55IU/L (normal <31), alkaline phosphatase level (ALP) of 428IU/L (normal <314) and gamma glutamyl transferase (GGT) of 345IU/L (normal <55 IU/L). Serum albumin, prothrombin time, viral markers for hepatitis B and hepatitis C were normal. Venous ammonia was a bit raised (195micromol/L, normal 9-33micromol/L). studies for pyruvate concentrations, serum thiamine level and red cell transketolase were not performed.
DWI (diffusion weighted image) and T2-FLAIR MRI of the brain showed multiple areas of white matter signal alteration involving both thalamo-capsulo-ganglionic and brainstem regions consistent with osmotic demyelination syndrome (Figure 1 & 2).
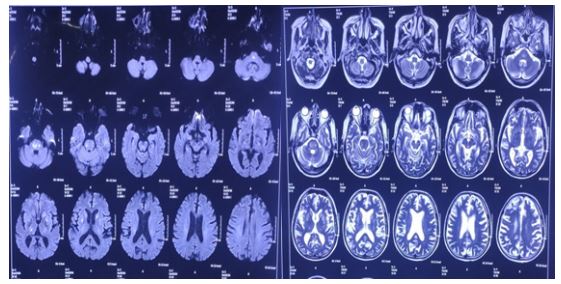
Figure 1&2: DWI(diffusion weighted image) and T2-FLAIR MRI of the brain showed multiple areas of white matter signal alteration involving both thalamo-capsulo-ganglionic and brainstem regions consistent with osmotic demyelination syndrome(ODS).
The constellation of symptoms (confusion, ataxia, and amnesia) in the setting of chronic alcohol use disorder led to a working diagnosis of Wernicke-Korsakoff Syndrome (WKS). However, MRI findings in WKS typically reveal abnormalities in the mammary bodies and periventricular regions. In our patient MRI of brain findings were consistent with ODS. On review of the literature, ODS on MRI of brain has been associated with WKS.
The patient was started treatment with intravenous thiamine (300mg/day). Fluid restriction to 1000ml/day and tolvaptan 15mg/day was instituted. The patient’s conscious level and ataxia improved. He also developed a diuresis as his plasma: urine osmolality ratio reversed. The serum sodium steadily rose to normal over 3 weeks while daily vitamin B therapy continued. Water restriction was abandoned after 72Hr and vaptan was continued for 10 days. At discharge 3 weeks after admission the patient had a slightly wide-based gait with some residual impairment of short-term memory and a tendency to confabulate. On discharge, Serum sodium was 135mmol/L, potassium was 4.5mmol/L.
Discussion
Wernicke [2] first addressed WE in 1881 as superior acute hemorrhagic poliencephalitis in two men with alcoholism and in a woman with pyloric stenosis. The association between thiamine deficiency and WE was first suspected in the 1940s [3]. Currently, thiamine deficiency plays a crucial role in the pathogenesis of WE. The prognosis of WE critically depends on the starting time of thiamine supplementation [4]. Thiamine is needed in the cell membrane to sustain osmotic gradients and is concerned in glucose metabolism and neurotransmitter synthesis. In healthy adults, thiamine requirement is influenced by the carbohydrate intake and the daily necessary dose is approximately 1-2mg. Because the body’s store of thiamine is only 30-50mg, the reserve would be completely depleted at 4-6 weeks after the lack of thiamine intake. Various clinical diseases or conditions disrupt the adequate absorption of thiamine, including chronic alcoholism, gastrointestinal surgery, hyperemesis gravidarum (HG), chemotherapy, systemic infectious diseases and malnutrition [4,9-12,15-20]. More specifically, low rates of thiamine absorption at the mucosal level, hepatic dysfunction and alcohol-induced hypermetabolism of thiamine may trigger chronic thiamine deficiency [16]. Thiamine-deficient cell membranes are unable to maintain osmotic gradients and result in the swelling of intra- and extracellular spaces. Pathological features have revealed edema, spongy degeneration of the neutrophil, neuron sparing, swelling of capillary endothelial cells and extravasation of erythrocytes [20]. In WE, the blood-brain barrier is damaged in the periventricular regions where thiamine-related glucose and oxidative metabolism are increased5. Previous studies of MRI suggested that typical topographic lesions of WE were located symmetrically in the thalami, mamillary bodies, tectal plate and periaqueductal area [17,18,20,21]. T2-hyperintense lesions were found uncommonly in the cerebellum, dentate nuclei and medulla oblongata [22-25]. The present patient also had no atrophy in the mammillary bodies and the cerebellar vermis.
Table 1: Clinical Presentation, Diagnostic Criteria, and Predictors of Favorable Outcome in Wernicke’s encephalopathy, Korsakoff Syndrome, and ODS.
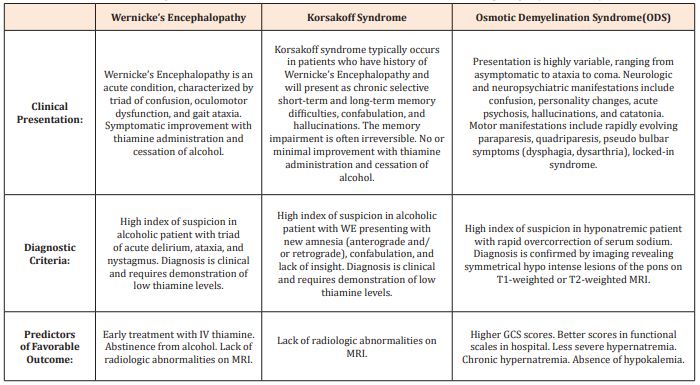
When WE is not treated, around 80% of patients will advance to the chronic form of the disease, Korsakoff Syndrome (KS). Clinical manifestations of KS include the classic WE triad as well as amnesia and confabulation [26]. KS generally carries a poorer prognosis compared to WE, as KS is typically irreversible despite treatment with thiamine and alcohol cessation. In contrast to patients with WE, patients with KS rarely recover and will require some form of social support.
The characteristic clinical picture in a vagrant with alcoholism, cortical atrophy on CT scan and a rapid improvement following B vitamin complex, was felt to be diagnostic of Wernicke’s encephalopathy, although studies for pyruvate concentrations and transketolase were not performed. The inappropriate urinary osmolality in the face of serum hypo-osmolality, the low urate [27], absence of fluid depletion with normal renal, thyroid, hepatic and adrenal function is considered good evidence for the syndrome of inappropriate antidiuretic hormone secretion. It has been suggested [13,14] that the SIADH might be expected to occur occasionally in Wernicke’s encephalopathy as does hypothermia [28,29] found the characteristic lesions in the supraoptic nuclei of the hypothalamus in 16-7% and in the paraventricular nucleus in 35% of their patients with Wernicke’s encephalopathy at post-mortem. The SIADH has been observed in various cerebral conditions and may present with confusion, fits and upgoing plantar responses [30]. It has been observed in hypothalamic lesions in particular, such as in acute intermittent porphyria with neuronal loss in the supraoptic and paraventricular nuclei [31], in neurohypophyseal choristoma [32] and in hypothalamic glioma [33]. Similarly the syndrome of inappropriate antidiuretic hormone secretion has been produced in goats by inducing lesions in the region of the supra-optic nuclei [34].
ODS is a demyelinating disorder of the brainstem and typically occurs with the rapid correction of serum sodium in hyponatremic patients [35]. As with WKS, ODS is also seen in the setting of nutritional and electrolyte stress, including chronic alcohol use disorder and malnourishment. The diagnosis of ODS is based on correlation of clinical findings with radiologic studies, in which an MRI reveals a symmetric area of demyelination. The treatment of ODS is usually supportive. Interestingly, the clinical presentation of CPM can vary from asymptomatic, to neuropsychiatric, to paralysis, coma, or death [36]. The neurological findings that were seen in our patient include disinhibiting, confusion, impaired cognition, and gait instability. It is important to note that it is difficult to distinguish the mild forms of ODS from WKS as both diseases present with similar neurological findings.
While ODS can vary from incidental finding to a fatal diagnosis, there are only a few studies that examine the prognostic indicators and functional outcomes. A retrospective cohort study of 25 patients with ODS demonstrated that higher GCS (Glasgow Coma Scale) scores (≥11), better scores in functional scales, less severe hyponatremia ([Na+] > 115mEq/L), and absence of superadded hypokalemia predict favorable outcome [37]. Another article reviews the homeostatic mechanism by which severe acute hyponatremia (serum [Na+] dropping to <120mEq/L in ≤48-hours) causes a more serious presentation compared to a slowly progressive chronic hyponatremia [38]. If untreated, severe acute hyponatremia may lead to brain edema, irreversible neurologic damage, respiratory arrest, brainstem herniation, and even death. In contrast, in chronic or less severe hyponatremia, symptoms usually do not arise until the serum [Na+] falls below 120mEq/L and they are initially nonspecific (e.g., headache, lethargy, nausea) [39]. Ultimately, however, the rate of sodium correction is more important than the nadir sodium concentration in the development of ODS.
Unlike WKS, where clinical outcome was favorable when fewer radiological signs were detected at onset, the extent of pontine signal abnormality in ODS does not predict clinical outcome. A retrospective cohort study of 24 patients with ODS concluded that the volume of demyelination on MRI was not associated with disease severity. This information is helpful and can prevent both premature and pessimistic prognostication based on radiologic abnormalities [40].
In patients with WKS complicated by ODS, as in our patient, it is especially important to consider the prognostic factors in the context of both WKS and ODS, and to assess the patient’s functional status prior to discharge. Using the prognostic factors for WKS we describe in this paper, our patient had only one favorable predictor in the context of WKS, which was immediate treatment with IV thiamine. It is also important to note that she had improvement in her short-term memory, a finding that is rare in patients with WKS. Additionally, the presence of MRI abnormalities is an unfavorable prognostic indicator for our patient in the context of WKS.
Using the prognostic factors of ODS we describe in this paper, our patient had several findings that would predict favorable outcome: no evidence of hyponatremia or hypokalemia, functional independence (ambulation without assistance), and GCS score of 13-14 throughout hospital stay.
Conclusion
Although hyponatraemia may give rise to neurological signs, an underlying neurological lesion causing the hyponatraemia should not be forgotten, least of all Wernicke’s encephalopathy where intravenous vitamin B complex may be life-saving. There are several cases in the literature of the SIADH where Wernicke’s encephalopathy was not apparently excluded. In addition to treating the patient for WKS with thiamine, ODS in the setting of WKS also necessitates assessment of the patient’s functional status. Only a few small studies exist that examine the predictors of disease severity and functional outcomes in WKS and ODS. There is very little literature about patients with both concurrent WKS and ODS as in our patient. This case is important because it highlights the subtle and overlapping differences between WKS and ODS from a clinical, diagnostic, and outcomes perspective.
References
- Latt N, Dore G (2014) Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J 44: 911-915.
- Wernicke C (1881) Die akute haANmorrhagische polioencephalitis superior. Fischer Verlag, Kassel. Lehrbuch der Gehirnkrankheiten fuANr AANrzte und Studierende II: 229-242.
- Campbell ACP, Russell WR (1941) Wernicke’s encephalopathy: the clinical features and their probable relationship to vitamin B deficiency. Q J Med 10(1): 41-64.
- Harper CG, Giles M, Finlay-Jones R (1986) Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry 49(4): 341-345.
- Harper C, Butterworth R. Nutritional and metabolic disorders. In: Greenfields Neuropathology. 6th ed. Graham DI, Lantos PL (Eds). Edward Arnold: UK; 1997: 601-652.
- Brunner JE, Redmond JM, Haggar AM, Stanton BE (1988) Central pontine myelinolysis after rapid correction of hyponatremia: a magnetic resonance study. Ann Neurol 23(4): 389-391.
- Gocht A, Colmant HJ (1987) Central pontine and extrapontine myelinolysis: a report of 58 cases. Clin Neuropathol 6(6): 262-270.
- Chason JL, Landers JW, Gonzalez JE (1964) Central pontine myelinolysis. J Neurol Neurosurg Psychiatry 27(9): 317-325.
- Bergin PS, Harvey P (1992) Wernicke’s encephalopathy and central pontine myelinolysis associated with hyperemesis gravidarum. BMJ 305(6852): 517-518.
- Peeters A, Van de Wyngaert F, Van Lierde M, Sindic CJM, Laterre EC (1993) Wernicke’s encephalopathy and central pontine myelinolysis induced by hyperemesis gravidarum. Acta Neurol Belg 93(5): 276-282.
- Olindo S, Smadja D, Cabre P, Mehdaoui H, Heinzlef O (1997) Gayet- Wernicke encephalopathy and centropontine myelinolysis induced by hyperemesis gravidarum. Rev Neurol (Paris) 153(6-7): 427-429.
- Falcone N, Compagnoni A, Meschini C, Perrone C, Nappo A (2003) Central pontine myelinolysis induced by hypophosphatemia following Wernicke’s encephalopathy. Neurol Sci 24(6): 407-410.
- Shalhoub RJ, Antoniou LD (1969) The mechanism of hyponatremia in pulmonary tuberculosis. Annals of Internal Medicine 70: 943.
- Ebels EJ (1978) How common is Wernicke-Korsakoff syndrome? Lancet 312(8093): 781-782.
- Gui QP, Zhao WQ, Wang LN (2006) Wernicke’s encephalopathy in nonalcoholic patients: clinical and pathologic features of three cases and literature reviewed. Neuropathology 26(3): 231-235.
- Thomson AD (2000) Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol Suppl 35(1): 2-7.
- Zuccoli G, Gallucci M, Capellades J, Regnicolo L, Tumiati B, et al. (2007) Wernicke encephalopathy: MR findings at clinical presentation in twenty-six alcoholic and nonalcoholic patients. AJNR Am J Neuroradiol 28(7): 1328-1331.
- Zuccoli G, Pipitone N (2009) Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol 192(2): 501-508.
- Haid RW, Gutmann L, Crosby TW (1982) Wernicke-Korsakoff encephalopathy after gastric plication. JAMA 247: 2566-2567.
- Suzuki S, Ichijo M, Fujii H, Matsuoka Y, Ogawa Y (1996) Acute Wernicke’s encephalopathy: comparison of magnetic resonance images and autopsy findings. Intern Med 35(10): 831-834.
- Fei G-q, Zhong C, Jin L, Zhang Y, Zheng X, et al. (2008) Clinical characteristics and MR imaging features of nonalcoholic Wernicke encephalopathy. AJNR Am J Neuroradiol 29(1): 163-169.
- Bae SJ, Lee HK, Lee JH, Choi CG, Suh DC (2001) Wernicke’s encephalopathy: atypical manifestation at MR imaging. AJNR Am J Neuroradiol 22(8): 1480-1482.
- Zuccoli G, Motti L (2008) Atypical Wernicke’s encephalopathy showing lesions in the cranial nerve nuclei and cerebellum. J Neuroimaging 18(2): 194-197.
- Liu YT, Fuh JL, Lirng JF, Li AF, Ho DM, et al. (2006) Correlation of magnetic resonance images with neuropathology in acute Wernicke’s encephalopathy. Clin Neurol Neurosurg 108(7): 682-687.
- Kang SY, Kang JH, Choi JC, Choi G (2005) Wernicke’s encephalopathy: unusual manifestation on MRI. J Neurol 252(12): 1550-1552.
- Arts NJ, Walvoort SJ, Kessels RP (2017) Korsakoff’s syndrome: A critical review. Neuropsychiatr Dis Treat 13: 2875-2890.
- Beck LH (1979) Hypouricemia in the syndrome of inappropriate secretion of antidiuretic hormone. New England Journal of Medicine 301(10): 528-530.
- Philip G, Smith JF (1973) Hypothermia and Wernicke’s encephalopathy. Lancet p. 121.
- Victor M, Adams RD, Collins GH (1971) The Wernicke-Korsakoff Syndrome. Blackwell Scientific Publications, Oxford.
- Bartter FC, Schwartz WB (1967) The syndrome of inappropriate secretion of antidiuretic hormone. American Journal of Medicine 42: 790.
- Perlroth MG, Tschudy DP, Marver HS, Berard CW, Zeigel RF, et al. (1966) Acute intermittent porphyria: new morphologic and biochemical findings. American Journal of Medicine 41(1): 149-162.
- Haslett C, Douglas NJ (1978) Inappropriate ADH secretion associated with neurohypophyseal choristoma. British Medical Journal 2(6154): 1753.
- Brisman R, Chuturian AM (1970) Inappropriate antidiuretic hormone (hypothalamic glioma in a child). Archives of Neurology (Chicago) 23(1): 63-69.
- Rundgren M, Fyhrquist F (1978) Transient water diuresis and syndrome of inappropriate antidiuretic hormone induced by forebrain lesions of different location. Acta physiologica scandinavica 103(4): 421-429.
- Mohammed A, Boddu P, Yazdani D (2016) Clinical evolution of central pontine myelinolysis in a patient with alcohol withdrawal: A blurred clinical horizon. Case Rep Med.
- King J, Rosner M (2010) Osmotic demyelination syndrome. The American Journal of the Medical Sciences 339(6): 561-567.
- Kallakatta R, Radhakrishnan, Fayaz R, Unnikrishnan J, Kesavadas C, et al. (2011) Clinical and functional outcome and factors predicting prognosis in osmotic demyelination syndrome (central pontine and/or extrapontine myelinolysis) in 25 patients. J Neurol Neurosurg Psychiatry 82(3): 326-331.
- Giuliani C, Peri A (2014) Effects of hyponatremia on the brain. J Clin Med 3(4): 1163-1177.
- Androgue H (2005) Consequences of inadequate management of hyponatremia. Am J Nephrol 25(3): 240-249.
- Graff-Radford J, Fugate J, Kaufmann T, Mandrekar J, Rabinstein A (2011) Clinical and radiologic correlations of central pontine myelinolysis syndrome. Mayo Clin Proc 86(11): 1063-1067.
